Focused library
Computer-based methods of focused library creation.
Silicon brain
Today, in silico drug design (CADD) is used by the vast majority of pharmaceutical leaders, including ChemDiv. Here we will give three examples of using CADD for the creation of focused libraries.
Recurring Neural Networks (RNNs)
One of the rapidly developing CADD methods involves RNNs, where, after training, the network generates output similar to the input, i.e. new molecular structures imitating the ones in the learning dataset.
In article [1], the automated focused library creation via transfer learning – i.e. training on a large set (of molecules, in this case, but the concept is not limited to them) first, and then tuning with smaller samples for lead optimization was explored.
After using a ChEMBL dataset to train an RNN, transfer sets that mimic those usually occurring in the medicinal chemistry workflow were selected.
Out of all the metrics chosen, two were key for evaluating the network’s performance: a unique-novelty score and a chemical closeness score.
Somewhat counterintuitively, smaller datasets required more training and larger ones were fine with fewer cycles. Lower fragment counts (meaning fewer distinct groups – fragments – were in the dataset) made for lower uniqueness, since the training data was more homogeneous.
These results are presented in this table (header numbers refer to completed epochs (i.e. cycles), cells to how much of the output, in percents, had a low (less than a quarter) unique-novelty score:
| Filename | Frag count | 5 | 10 | 12 | 15 | 17 | 20 |
| DHODH full | 66 | -- | 1 | 59 | 91 | 96 | 100 |
| METAP2 full | 59 | -- | 60 | 78 | 88 | 91 | 100 |
| MMP-12 full | 31 | 33 | 66 | 80 | 94 | 99 | 100 |
| P2X7 full | 131 | -- | -- | -- | 18 | 78 | 99 |
| SLC22A12 full | 49 | -- | 75 | 83 | 98 | 100 | 100 |
| DHODH subset | 41 | -- | 46 | 62 | 88 | 98 | 100 |
| METAP2 subset | 40 | -- | 60 | 76 | 92 | 100 | 100 |
| MMP-12 subset | 22 | 50 | 80 | 87 | 97 | 100 | 100 |
| P2X7 subset | 64 | -- | 34 | 85 | 95 | 99 | 100 |
| SLC22A12 subset | 32 | 13 | 75 | 88 | 100 | 100 | 100 |
| US-20090018134 | 33 | 8 | 58 | 79 | 91 | 93 | 99 |
| US-20090286778 | 123 | -- | 21 | 55 | 75 | 81 | 83 |
| US-20100016279 | 73 | -- | 82 | 97 | 99 | 100 | 100 |
| US-20120157425 | 91 | 1 | 85 | 92 | 99 | 100 | 100 |
| WO-2010079443 | 54 | -- | -- | -- | 8 | 60 | 92 |
| WO-2011075515 | 137 | -- | 2 | 42 | 89 | 93 | 100 |
| WO-2012053186 | 44 | 1 | 66 | 87 | 94 | 100 | 100 |
| WO-2012067965 | 110 | -- | 34 | 85 | 97 | 98 | 100 |
SIFt
Another common technique in CADD is SBF (structure-based focusing), in which specific interaction constraints are used as the basis to design new chemical compounds that could bind to the target.
In article [2], researchers developed a method for large scale data analysis and visualization – structural information fingerprint (SIFt). In order to leverage the three-dimensional nature of the molecules more effectively, r-SIFt was developed, with 'r' referring to different R groups.
After assembling virtual libraries and docking poses, two-dimensional descriptors were found via Pipeline Pilot, at which point r-SIFts were generated, integrating the binding parameters into the fingerprint. For the 10 poses with the highest Cscores (for MAP kinase p38 inhibitors), r-SIFts were subsequently generated, with the best pose selected through calculating the Tanimoto coefficient.
The results were evaluated by measuring the predictive accuracies of the decision trees made using the r-SIFTs produced previously.
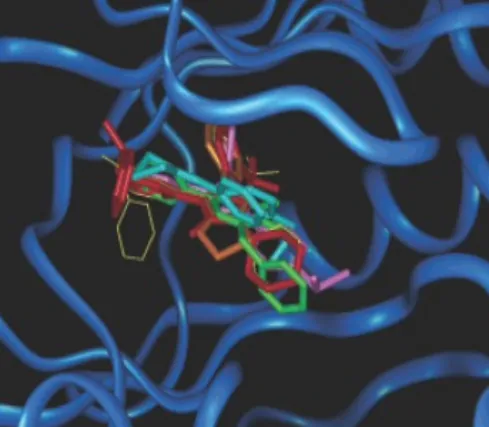
Combined with a conventional toolkit, r-SIFt proved to be a great tool for visualization that zoomed in on particular parts of the molecule. The following figure shows the ways in which p38 inhibitors are alike and, upon further inspection, reveals the differences.

b is an overlay of the best docking pose (c-f are p38 inhibitors, g is not). The cocrystal structure of c is shown with a yellow line. Inhibitors bind in a similar way: purple parts are near the hinge, the blue ones are concentrated in the hydrophobic pocket.

Structures and R groups. 1-5 correspond to c-g in the previous picture.
findings. A trifluorobenzene R1 of 1 compared to smaller 3-fluorophenol R1 explains the higher degree of interaction in the hydrophobic region.
Multiobjective genetic algorithm
A multiobjective genetic algorithm (MOGA) was employed as a foundation for MoSELECT – a program that searches the virtual space for solutions and presents the connections between different targets [3].
Tasks with many objectives frequently have different lines of solutions, each of them having different trade-offs. A standard genetic algorithm searches these lines separately, unlike MOGA, which does so simultaneously, utilizing the idea of 'dominance':

The task is to minimize f1 and f2. Solid circles are for non-dominated answers, meaning there are no better solutions for both goals. Empty dots are dominated, with number showing how many 'dominators' – better solutions – are present.
When tasked with creating a focused library for a random molecule from 2-Aminothiazole library, optimizing for similarity (measured by Daylight fingerprints and the Tanimoto coefficient) and cost, SELECT – which used a standard genetic algorithm – provided only a unilaterally adequate solution – either the averages 0,832; US$48 289,4 or 0,696; 1 675,2. The only way to achieve a compromise -- painstakingly choosing weights -- is hard for such non-commensurate goals. MoSELECT, instead of giving single solutions, creates the entire family of non-dominated answers and allows for an easier choice in deciding on the compromise:

The expanded version of the third figure, the entire family of solutions is shown.
Conclusion
ChemDiv offers first-class CADD services in the field of cheminformatics, which include virtual screening, docking, hit2lead optimization and others.
Literature
[1] Guidelines for RNN Transfer Learning Based Molecular Generation of Focused Libraries; Amabilino et al., Journal of Chemical Information and Modeling 2020, 60, 12, 5699–5713
[2] Knowledge-Based Design of Target-Focused Libraries Using Protein - Ligand Interaction


