Screening compounds

Screening compounds: looking for a needle in a haystack
One of the early stages of drug discovery is screening the compounds available – be it a virtual screening or high-throughput screening. This stage is necessary for hit identification, which, later in the drug design pipeline will become leads, which, maybe, will become a drug after a dozen or so years. In this short piece about screening compounds, we will cover some virtual screening techniques as well as some HTS assays used for compound screening at ChemDiv.
Virtual screening: automating common sense
In silico approach allows to work through the screening compounds in a very time-efficient manner by both utilizing computational chemistry and some “common sense” rules medicinal chemists use: like excluding the scaffolds that commonly cause issues down the line due to giving false positives (PAINS), for example. Common approaches here include docking and calculating drug-likeness with characteristics like the tanimoto coefficient, which tells how chemically similar the molecules are, allowing for a quick comparison.
Docking, in a somewhat simplified summary, entails answering a somewhat simple question: does the ligand actually fit the target? A numerical answer to that question is the binding energy, the lower, the better. Programs will estimate it through methods (the evolutionary approach may serve as an example) which give a more-or-less correct answer instead of a more precise one, sacrificing accuracy for speed, which is more important when dealing with the amount of screening compounds tested at the early stages. Moreover, since the virtual screening does not require actual compounds, it is possible to test a larger subset of the chemical space than one has access to.
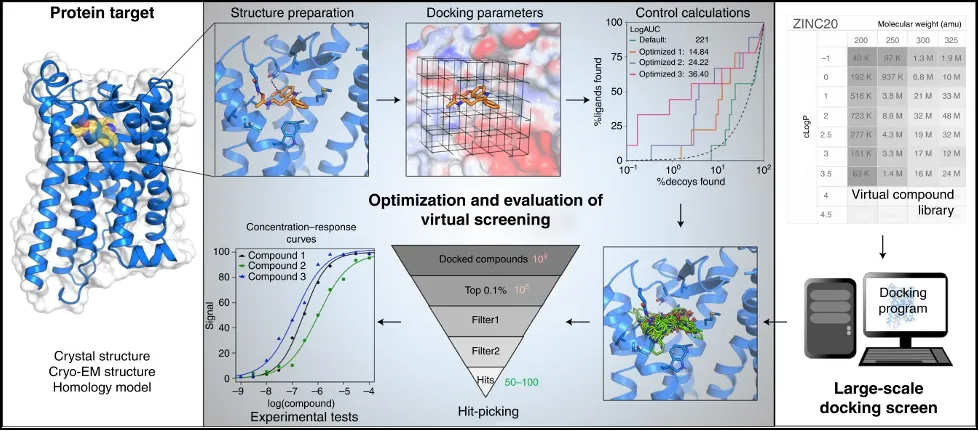
Virtual screening plan, from “A practical guide to large-scale docking” by Bender et al.
While it would be tempting to rank the molecules and just send the top 5% percent for HTS, the reality is that the work has just started. Because the calculations are optimized for speed, they are not used to rank molecules. Additionally, screening compounds should be filtered for certain interactions with the target, further lowering the amount of molecules that need to be tested.
High-throughput screening: diagonal reading
After selecting screening compounds from the results of the virtual screening, the next stage in the pipeline is the high-throughput screening. If, at the previous stage, we sampled the chemical space for molecules that might have some activity, now is the time to actually check them in a variety of activity-measuring assays, which include, but are not limited to:
TR-FRET, a fluorescence assay
ADP Hunter, which gives a fluorescent signal proportional to the amount of ADP
Z’-Lyte, in which peptides give a signal, if they were protected by a kinase
pNA assays, in which yellow-colored p-nitroaniline serves as a measure of activity ● ELISA, an antigen-antibody assay
All of these assays, and many more, are used by ChemDiv to work through the screening compounds and deliver the best results possible.
Still, just like virtual screening, HTS is built for speed, which means there are some sacrifices when it comes to accuracy. The more promising hits become leads, at which point one might say that, even if the needle has not been found in the haystack of chemical space, something sufficiently similar has.
Conclusion
Screening compounds go through a variety of screens to minimize costs while maximizing benefits: nowadays the process of screening starts with the virtual screening, which answer the more fundamental questions quickly, but with a somewhat low accuracy. After that, the more promising molecules go through numerous assays, depending on the nature of their target. This allows us to efficiently throw away the molecules that do not demonstrate desired activity and select the few that, if we are lucky, eventually end up as a drug on the shelves years after.


